Lorne Proteins 2020


Lorne Proteins 2020
If you’ve landed here after checking out my poster @LorneProteins, welcome. If you stumbled here by accident, the more the merrier. Either way, thanks for stopping by!
I hope you found something interesting or useful, and enjoyed hearing about the work we are doing to develop new tools for measuring proteostasis. If you have any questions, want to know more about what we do, or simply want to get in touch, you can find me on twitter @dezeraecox head over to the contact page.
For anyone who wasn’t at the conference, or didn’t get a chance to say hi, I have included a brief run-down (and tried to skip over most of the gory – boring, technical – details!).
What’s all this proteostasis business?
Cells have an extensive quality control network responsible for maintaining their molecular machines, including synthesis, folding, degradation and transport
\[1\]. Collectively, this machinery is known as the proteostasis network. Proteostasis imbalance results in protein misfolding and aggregation, the central molecular signature of neurodegenerative diseases such as Alzheimer’s and Parkinson’s.
We lack knowledge of which proteins in the cell become vulnerable to improper folding during proteostasis imbalance. One measure of protein foldedness is the extent to which a protein can be unfolded thermally or with a chemical denaturant such as urea. Until recently, it has been difficult to track foldedness in cells due to sheer complexity of the many ten’s of thousands of proteins and proteoforms needed for our cells to function.
To overcome this, this work specifically aimed to:
- Devise a chemical biology and proteomics approach to monitor the foldedness of the proteome.
- Determine the influence of pharmacological agents that unbalance proteostasis on proteome foldedness.
What is tetraphenylethene maleimide, other than a tongue twister?!
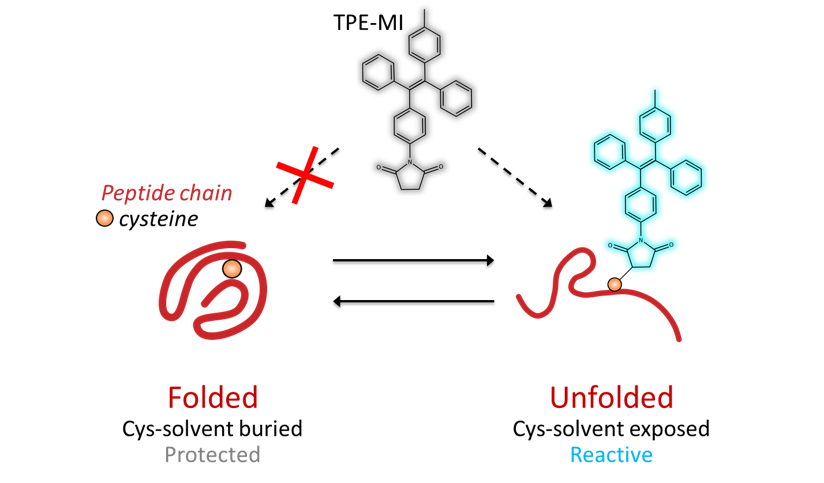
Tetraphenylethene maleimide (TPE-MI) is a dye (tetraphenylethene maleimide) that becomes fluorescent when it binds to reactive thiol residues
\[2\]. Buried, reactive thiols are the most buried residue of all amino acids in globular (properly folded) proteins. Monitoring the exposure of these thiols using TPE-MI gives us a sensitive way to probe foldedness in complex mixtures.
To test this strategy, we used a purified, well-studied protein – β-lactoglobulin. We know both the sequence and 3D structure of this protein, and its behaviour in a range of denaturants is known. β-lactoglobulin has 5 thiol residues, of which two pairs are bonded and the remaining one (Cys121) is buried in the core of the folded protein. As β-lactoglobulin unfolds in increasing concentrations of denaturant, we get a corresponding increase in TPE-MI fluorescence.
But what about the giant protein soup our cells make, I hear you ask…
While TPE-MI can reliably tell us about the unfolding of a single, purified protein, our cells are jam-packed with thousands of copies of thousands of different proteins. How can we possibly know which proteins TPE-MI is binding to?
Enter: proteomics. This revolutionary tool is a workflow based on mass spectrometry which can report back on the composition of complex mixtures – both which proteins are present, and the relative amount of that protein. The basics of this technique are beyond the scope of this post, but if you are looking for more details check out the resources section below
\[3-5\]. I have included a brief explanation of the method we use below, which relies on isotopically labelling cells in culture before lysing, denaturing and labelling with TPE-MI. Proteins are digested (chopped up) into regular, smaller pieces before analysis via mass spectrometry.
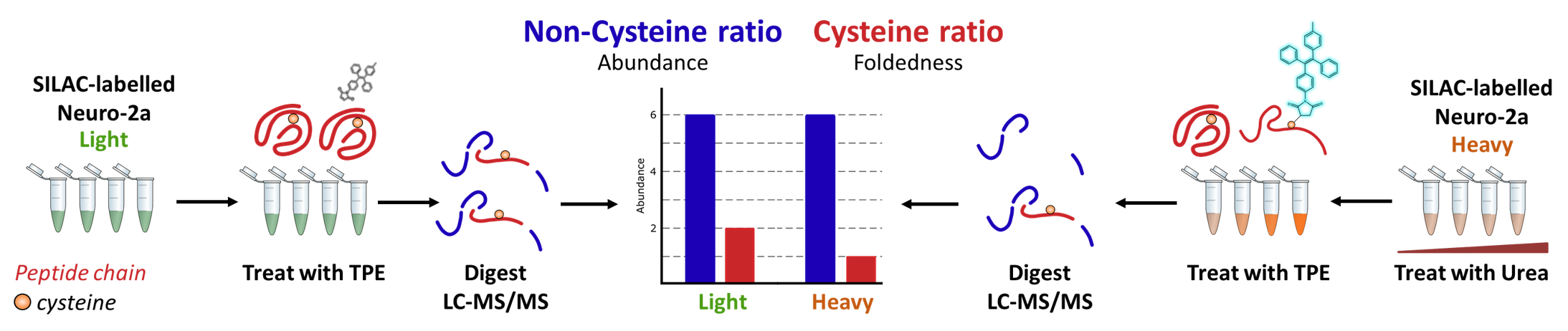
Using custom software, we can identify and quantify protein pieces. Unfortunately, we cannot identify the protein pieces specifically labelled with TPE-MI, but instead, we look for the loss of those pieces. We can do this for many many proteins and start to understand the concentration at which specific proteins unfold. We can even start to understand intricacies in unfolding for separate protein domains.
How does this help us understand disease mechanisms?
This new method now provides a way to monitor how the stability of proteins (measured how much denaturant it takes to unfold them) changes under conditions of proteostasis impairment that are present in disease. For example, we are now investigating the changes in proteome foldedness when we inhibit hubs of the proteostasis network such as molecular chaperones. How do the proteins that rely on specific molecular chaperones to be folded change when those proteins are no longer active? What if we stop the cell from degrading old proteins? Or from decorating their proteins with extra modifications that mediate structure and activity like phosphorylation? Defects in this machinery are common in neurodegenerative protein aggregation diseases and we now have the tools to start tackling these questions.
References and handy links
- Chiti F, Dobson CM (2017) Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade. Annu Rev Biochem 86: 27–68.
- Chen MZ, Moily NS, Bridgford JL, Wood RJ, Radwan M, Smith TA, Song Z, Tang BZ, Tilley L, Xu X, Reid GE, Pouladi MA, Hong Y, Hatters DM (2017) A thiol probe for measuring unfolded protein load and proteostasis in cells. Nat Commun 8: 1–10.
- Proteomics: Everything you always wanted to know but were afraid to ask.
- Lottspeich, F. (2009). Introduction to proteomics. In Proteomics (pp. 3-10). Humana Press.
- Graves, P. R., & Haystead, T. A. (2002). Molecular biologist’s guide to proteomics. Microbiology and molecular biology reviews, 66(1), 39-63.
That’s all from me for now. Still can’t get enough or want to know more? Simply want to get in touch? Find me on twitter @dezeraecox head over to the contact page - love to hear from you! Now, back to the beach 🏄🌴☀️ !